The Role of Tau Aggregation in Alzheimer’s Disease Pathology and Prevention
In the neurobiology landscape, the Tau protein plays a crucial role in stabilising microtubules within neurons. Positioned primarily in axons but also present in dendrites and various neuronal components, Tau undergoes significant transformations in certain neurodegenerative conditions known as tauopathies, where it separates from microtubules and forms insoluble aggregates in the brain.
Tau’s Connection to Alzheimer’s Disease
Alzheimer’s disease (AD), is the most prevalent neurodegenerative disease affecting approximately 10% of seniors over 65, according to data from the not-for-profit Alzheimer’s Association®. First identified by German scientist Alois Alzheimer in 1907, AD is characterized by the presence of amyloid beta plaques and neurofibrillary tangles (NFTs) containing Tau.
Tau Isoforms and Tauopathies
Tau is a protein that is encoded by the MAPT gene on human chromosome 17 and has four regions: the N-terminal, the proline-rich, the repeat, and the C-terminal. Tau has no well-defined structures and is highly soluble and hydrophilic. It can adopt a “paper-clip” conformation in the cytoplasm, where the N and C terminals are close together. Tau has six isoforms in the adult human brain, which differ in the number of microtubule binding repeat motifs (3R or 4R). These isoforms are generated by alternative splicing of the tau gene and vary in size and amino acid composition. In the fetal human brain, only the shortest isoform is expressed. Tau is involved in several neurodegenerative diseases, collectively known as tauopathies, where it forms aggregates called neurofibrillary tangles (NFTs). These diseases include Alzheimer’s disease, frontotemporal dementia, progressive supranuclear palsy, and others. Tau also accumulates in rod-like deposits in Huntington’s disease.
Tau Aggregation
Post-Translational Modifications
Tau exhibits a diverse range of post-translational modifications (PTMs), such as phosphorylation, glycation, nitration, O-GlcNAcylation, acetylation, oxidation, ubiquitination, sumoylation, and methylation. These PTMs intricately regulate tau’s affinity for microtubules, with elevated levels of ubiquitinated tau observed in Alzheimer’s disease (AD) and other tauopathies. Notably, lysine acetylation and other PTMs hinder tau function and contribute to aggregation.
Detection of acetylation at K280 in transgenic mouse models of tauopathies, absent in control mouse brains, suggests its potential association with tau pathology. Among the myriad post-translational modifications, phosphorylation stands out as the most extensively studied.
Tau Hyperphosphorylation
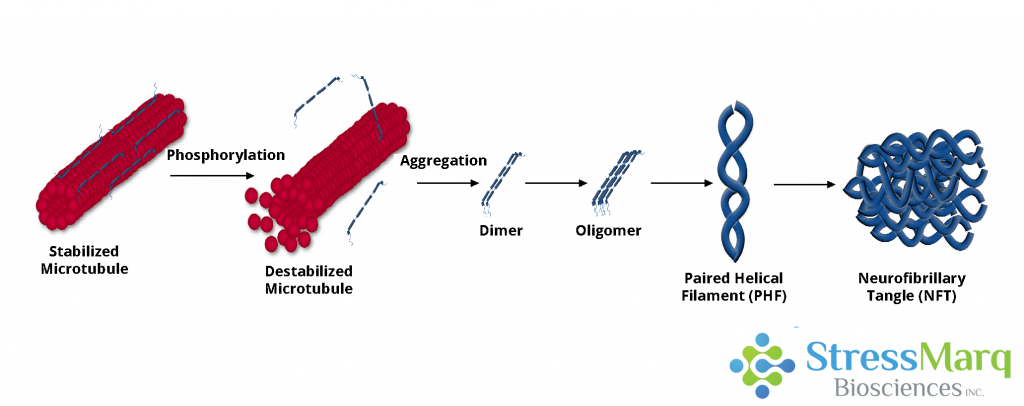
Approximately twenty percent of tau’s amino acids can undergo phosphorylation. Normally positively charged, the microtubule-binding domain of tau is attracted to microtubules. However, hyperphosphorylation alters this charge, causing tau to dissociate from microtubules, impeding its ability to assemble or stabilize them. The ensuing neurodegeneration may result from compromised microtubule function, tau’s toxicity to neurons, or a combination of these factors.
Paired Helical Filaments
Two hyperphosphorylated tau monomers can combine to form dimers, with interactions between hexapeptides in repeats 2 and 3 facilitating dimerization and subsequent oligomerization. Oligomers aggregate into Paired Helical Filaments (PHFs), characterized by a twisted double-helical ribbon structure. Tau within PHFs becomes more negatively charged, losing its ability to bind tubulin or effectively stabilize microtubules. The decay of the microtubule network leads to impaired neuronal function.
Neurofibrillary Tangles
Neurofibrillary Tangles (NFTs) represent filamentous, insoluble aggregates of tau, featuring an increased beta-sheet structure and composed of aggregated PHFs. The extent of NFT deposition correlates with dementia severity and neuron death. Other forms of tau aggregates in AD patients, such as neuropil threads and neuritic plaques, also contribute to neuron degeneration. Nevertheless, some research suggests that filamentous and fibrillary tau may exhibit neuroprotective effects, highlighting the complexity of tau’s role, while soluble, hyperphosphorylated tau oligomers emerge as the most toxic form.
Insights into Tau Propagation Mechanisms
Tau Propagation
One proposed mechanism driving the propagation of tau pathology is termed “prion-like propagation.” This process involves the transmission of tau ‘seeds’ or fibrils from a donor cell to a recipient cell, where they attract endogenous tau proteins to their ends. While the spread of tau fibrils mirrors prion-like characteristics, uncertainties persist regarding whether tau shares additional properties with prions, such as the potential for human-to-human transmission.
Tau Preformed Fibrils
Preformed fibrils (PFFs) possess the capability to induce tau aggregation in both cultured cells and living animals by enlisting soluble monomers to form insoluble fibrils. The introduction of minute quantities of tau PFFs into tau-expressing cells can prompt the recruitment of substantial tau amounts into “filamentous inclusions resembling NFTs.” Synthetic tau PFFs also exhibit the ability to enter non-neuronal cells and induce the recruitment of tau into NFTs.
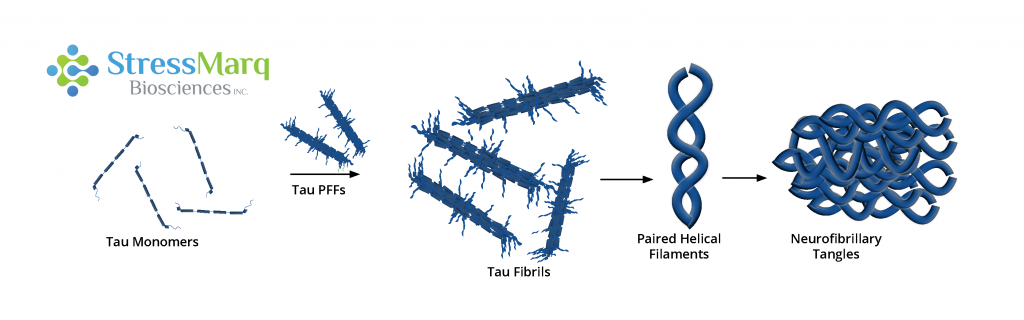
Tau Truncation
Certain studies indicate that full-length tau preformed fibrils (PFFs) exhibit a more efficient seeding of aggregation in primary neurons compared to truncated K18 tau. However, the co-expression of truncated and full-length tau has been demonstrated to induce severe yet reversible neurotoxicity in mice. This phenomenon might be attributed to the creation of soluble, non-filamentous, high-molecular weight oligomers that possess toxicity towards neurons.
Tau and Alpha Synuclein Interactions
More than 50% of AD cases involve the formation of Lewy bodies, and amyloid-beta plaques and NFTs are often found in the brains of patients with Parkinson’s Disease and Dementia with Lewy bodies. In vitro, alpha synuclein and tau mutually enhance each other’s fibrillization, and within Lewy bodies, tau fibrils co-localize with alpha synuclein fibrils. Additionally, alpha synuclein preformed fibrils (PFFs) contribute to tau phosphorylation and can prompt tau aggregation in vitro. The interplay and fibrillization of tau and alpha synuclein may occur in both tauopathies and synucleinopathies.
Therapeutic Approaches
Numerous potential treatments for AD and other tauopathies focus on targeting the tau protein. Addressing tau hyperphosphorylation, regulated by protein kinases and phosphatases, may involve inhibiting tau kinases, restoring protein phosphatase 2A, or targeting tau’s O-glycosylation. Another viable approach is targeting neuroinflammation, given its role in tau propagation. Immunizing mice with tau antibodies has demonstrated a slowing of disease progression, suggesting that immunotherapy holds promise as a treatment option.
Featured Tau Products
StressMarq offers a variety of tau monomers, oligomers, and pre-formed fibrils and antibodies for Alzheimer’s disease research.
Original Source: StressMarq Biosciences Inc. – Author: Patricia Thomson
Related Products