Amyloid Hypothesis vs Tau Hypothesis in Alzheimer’s Disease
In a recent scientific commentary, Brenda Knox at StressMarq delves into the complex interplay between amyloid-beta plaques and tau neurofibrillary tangles in Alzheimer’s disease. This commentary explores the historical context, analyzes the Amyloid Hypothesis and Tau Hypothesis, and delves into the complexities of tau-focused therapies. Going beyond these hallmarks, Knox also examines alternative disease mechanisms, offering essential insights into neurodegenerative research.
Plaques, Tangles, and the Pathology of Alzheimer’s Disease
As our global population ages, Alzheimer’s Disease (AD) has emerged as a pressing public health concern. Although the elusive cure for this neurodegenerative condition remains undiscovered, more than a century of research has offered insights into the disease’s progression and contributing factors. Central to this understanding are the two hallmark features found in AD-afflicted brains: amyloid-beta plaques and tau neurofibrillary tangles. While both amyloid-beta and tau aggregation are implicated in the disease, questions persist regarding the sequence of aggregation, the optimal treatment targets, and the intricate interplay between these two proteins.
A Glimpse into Alzheimer’s Disease Research History
Alois Alzheimer’s groundbreaking work in the early 20th century laid the foundation for our understanding of AD. In his detailed observations of Auguste Deter, a 51-year-old dementia patient, Alzheimer noticed symptoms like short-term memory loss and delusions typically associated with older patients, not those in their 50s. After her passing, he examined her brain and discovered the presence of plaques and tangles, with the latter being a unique finding. This pivotal discovery led to the naming of the disease we now know as Alzheimer’s.
In the 1930s and 40s, cases of autosomal dominant or early-onset familial AD garnered attention. These cases, linked to the inheritance of abnormal genes, manifested in patients under 65 and differed from the more common “senile dementia” observed in those over 65 without such inherited genetic traits.
The 1960s and 70s brought about a realization that the pathology of early-onset AD was akin to “senile dementia.” This transformed Alzheimer’s from a rare genetic disorder to a widespread public health concern, sparking a surge of interest and research funding.
The Amyloid Hypothesis
The amyloid hypothesis, also known as the amyloid-beta hypothesis or the amyloid cascade hypothesis, gained prominence following observations of individuals with Down syndrome who experienced neurological changes and memory loss later in life. These individuals exhibited plaque formation similar to that seen in AD patients. In 1984, Glenner and Wong isolated a 42 kDa peptide, amyloid-beta (Aβ), from the plaques in both AD and Down syndrome brains. Notably, they proposed a shared pathogenic process between AD and Down syndrome, given their common Aβ content. Furthermore, their belief that Aβ, encoded on chromosome 21, was responsible for AD pathology due to the trisomy of chromosome 21 in Down syndrome laid the foundation for the amyloid hypothesis. This hypothesis posits that Aβ aggregation initiates a cascade of events leading to AD pathology and symptoms. These events include the aggregation of hyperphosphorylated tau proteins into neurofibrillary tangles (NFTs), alongside inflammation and oxidative stress causing neuronal dysfunction and death.
The simplicity and therapeutic potential of the amyloid hypothesis made it a focal point of extensive research for several decades, despite the complex and conflicting theories surrounding AD’s pathogenesis.
The Tau Hypothesis: Unraveling the Intricacies of Neurodegeneration
In recent years, the Tau hypothesis has emerged as a prominent contender, gaining recognition for its contributions to our understanding of Alzheimer’s disease. Tau phosphorylation, it suggests, diminishes its capacity to facilitate microtubule assembly. This impairment results in neurodegeneration through synaptic dysfunction and neuronal loss. Moreover, neurofibrillary tangles, composed of tau, can induce neuronal dysfunction and death. An interesting aspect is that tau tangles have been observed in the brains of individuals with mild dementia, even in the absence of Aβ pathology. The correlation between tau pathology and AD progression is stronger than that of Aβ plaque load, further underscoring the significance of tau in the disease.
While Aβ plaques have been identified in individuals without neurodegeneration, the presence of NFTs, often associated with frontotemporal dementia and other tauopathies, paints a different picture. This disparity can be attributed to the location of these aggregates: Aβ plaques exist in the extracellular space, while tau tangles form within neurons, severely impeding axonal transport.
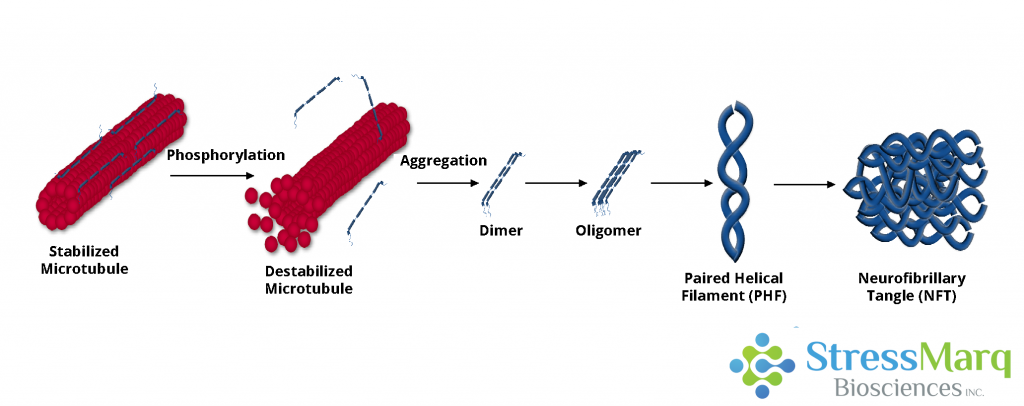
Although tau research has historically been overshadowed by the dominance of the amyloid hypothesis, recent developments have breathed new life into the exploration of tau-focused strategies. Seven anti-tau therapies are currently progressing through phase II trials, offering a glimmer of hope in the search for effective AD treatments.
Challenges and Controversies in Tau Research
Despite the promise of anti-tau therapies, several have faltered in clinical trials. One such example is Tideglusib, a GSK-3β inhibitor, which showed limited clinical benefit in a phase II trial. The Methylene blue dye derivatives Trx0014 and LMTM, which inhibit tau aggregation, seemed to slow cognitive decline in phase III trials. However, concerns linger about the methodology and the validity of claims regarding their efficacy.
A Joint Mechanism: Exploring the Amyloid-Tau Connection
Rather than viewing the tau and amyloid hypotheses as rivals, it’s increasingly evident that AD is a multifaceted disorder necessitating a multifaceted approach to treatment. The interaction between Aβ and tau in triggering AD pathology has been likened to various metaphors, from a “toxic pas de deux” to a “trigger and a bullet.” Most researchers propose that Aβ initiates tau tangle formation, subsequently harming neurons. Various mechanisms, such as Aβ-induced inflammation, synaptic defects promoting tau aggregation, Aβ cross-seeding of tau, and even the potential for a feedback loop where tau mediates Aβ toxicity, are all under scrutiny.
Beyond Plaques and Tangles: Exploring Alternate Mechanisms
AD’s complexity extends beyond amyloid plaques and tau tangles. Oxidative stress and inflammation also play pivotal roles in the disease’s progression. The mitochondrial cascade hypothesis proposes that impaired mitochondrial function, influenced by maternal genetics, sets in motion the aggregation of tau and Aβ. Intriguingly, herpes simplex virus type 1 (HSV1) has been associated with an increased risk of AD in APOE-ε4 carriers, suggesting a potential role for viruses and other microbes in the disease’s etiology.
Additionally, the cholinergic hypothesis posits that acetylcholine (ACh) dysfunction contributes to AD by inducing inflammation and promoting Aβ and tau aggregation. This theory underscores the significance of ACh in AD, as four acetylcholinesterase inhibitors are currently employed to alleviate symptoms in patients. However, these medications provide symptom relief rather than a cure.
In conclusion, Alzheimer’s pathology is a multifaceted puzzle that continues to challenge researchers and medical professionals. Finding a cure may not be as straightforward as initially hoped, and alternative approaches, such as prophylactic measures and multi-pronged treatment strategies, may be necessary. With notable investments in AD research, such as Bill Gates’ recent $100 million donation, and ongoing advancements in laboratories worldwide, the journey towards understanding and combating this devastating disease remains a beacon of hope.
StressMarq: Aiding Alzheimer’s Research
For researchers immersed in the study of Alzheimer’s, Parkinson’s, and other neurodegenerative diseases, StressMarq offers a range of innovative products designed to support your work. StressMarq’s recent launch of Aβ monomers, oligomers, and pre-formed fibrils, coupled with their diverse selection of tau fibrilized proteins and Aβ and tau antibodies, represents a valuable asset in the quest to decipher the mysteries of these debilitating conditions. Their strategic focus on providing pathology-inducing fibrilized proteins and associated derivatives aligns seamlessly with the growing demand for advanced tools in neurodegenerative disease research.
To find out more please visit: www.stressmarq.com
Original Source: Amyloid Hypothesis vs Tau Hypothesis by Brenda Knox